Ask Ethan: How Well Has Cosmic Inflation Been Verified?
Some claim that inflation isn’t science, but it sure has made some incredibly successful scientific predictions.
So, you want to know how the Universe began? You’re not alone. Every other curious member of humanity, for as long as recorded history exists (and probably much longer), has wondered about exactly this question, “where does all this come from?” In the 20th century, science advanced to the point where a large suite of evidence pointed to a singular answer: the hot Big Bang.
Yet a number of puzzles arose that the Big Bang was unable to solve, and a theoretical add-on to the Big Bang was proposed as the ultimate cosmic solution: inflation. This December will mark 40 years since inflation was proposed by Alan Guth, and Paul Erlich wants to know how well inflation has stood the test of time, asking:
To what margin of error or what level of statistical significance would you say you say inflation has been verified?
The short answer is “better than most people think.” The long answer is even more compelling.
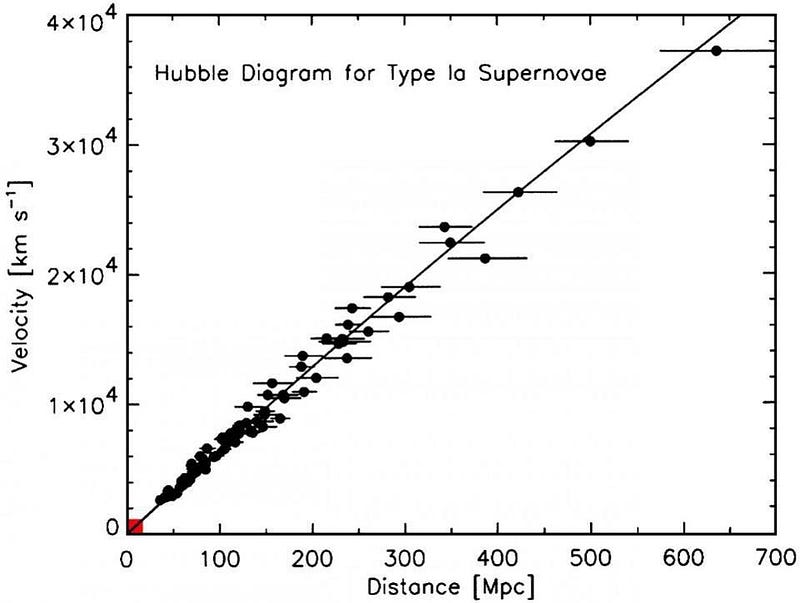
The Big Bang is an incredibly successful theory. It began from just two simple starting points, and made an extrapolation from there. First, it insisted that the Universe be consistent with General Relativity, and that is the theory of gravity that we should use as our framework for building any realistic model of the Universe. Second, it demanded that we take seriously the astronomical observations that galaxies, on average, appear to be receding from us with speeds that are in direct proportion to their distance from us.
The simplest way to proceed is to let the data guide you. In the context of General Relativity, if you allow the Universe to be evenly (or roughly evenly) filled with matter, radiation, or other forms of energy, it will not remain static, but must either expand or contract. The observed redshift-distance relation can be directly explained if the fabric of space itself is expanding as time goes on.

If this is the picture of the Universe you put together, it can carry some enormous consequences along for the ride. As the Universe expands, the total number of particles within it remains the same, but the volume increases. As a result, it gets less dense. Gravity pulls things into progressively larger-scale clumps with the passage of more time. And radiation — whose energy is defined by its wavelength — sees its wavelength stretch as the Universe expands; hence, it becomes cooler in temperature and lower in energy.
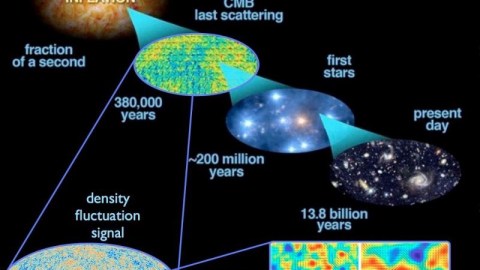
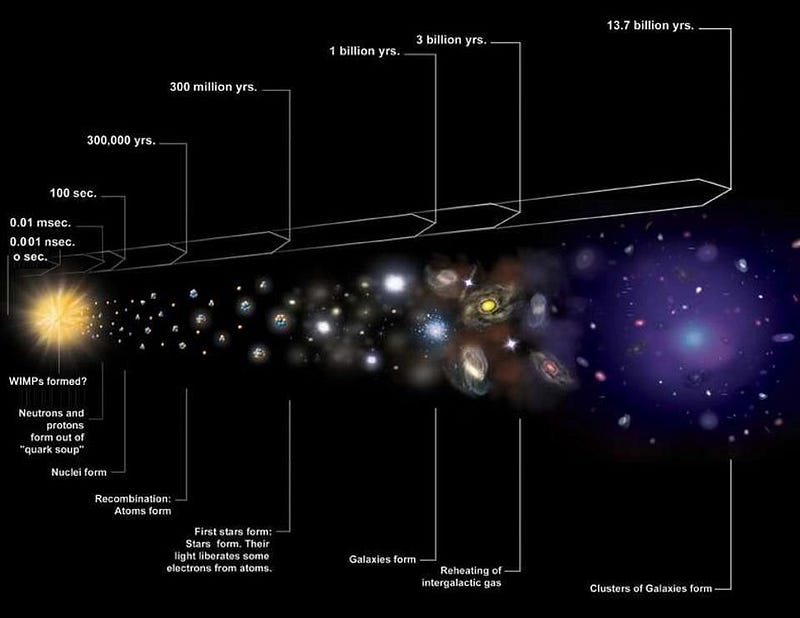
The huge idea of the Big Bang is to extrapolate this idea backwards in time, to higher energies, higher temperatures, greater densities, and a more uniform state.

This led to three new predictions, in addition to the expanding Universe (which had already been observed). They were as follows:
- The earliest, hottest, densest times should allow for a period of nuclear fusion early on, predicting a specific set of abundance ratios for the lightest elements and isotopes even before the first stars form.
- As the Universe cools further, it should form neutral atoms for the first time, with the leftover radiation from those early times traveling unimpeded and continuing to redshift until the present, where it should be just a few degrees above absolute zero.
- And finally, whatever initial density imperfections are present should grow into a vast cosmic web of stars, galaxies, galaxy clusters, and cosmic voids separating them over the billions of years that have passed since those early stages.
All three predictions have been verified, and that’s why the Big Bang stands alone among theories of the Universe’s origins.
But that doesn’t mean the Big Bang explains everything. If you extrapolate all the way back to arbitrarily high temperatures and densities — all the way back to a singularity — you wind up with a number of predictions that don’t pan out in reality.
We don’t see a Universe with different temperatures in different directions. But we should, since a region of space tens of billions of light-years to your left and another one tens of billion of light-years to your right should never have had time to exchange information since the Big Bang.
We don’t see a Universe with leftover particles that are relics from some arbitrarily hot time, like magnetic monopoles, despite the fact that they should have been produced in great abundance.
And we don’t see a Universe with any measurable degree of spatial curvature, despite the fact that the Big Bang has no mechanism to exactly balance energy density and spatial curvature from an extremely early time.
The Big Bang, on its own, offers no solution to these puzzles. It succeeds if we extrapolate back to a hot, dense, almost-perfectly-uniform early state, but it doesn’t explain any more than that. To go beyond these limitations requires a new scientific idea that supersedes the Big Bang.
But superseding the Big Bang isn’t easy at all. To do so, a new theory would have to do all three of the following:
- Reproduce all of the successes of the Big Bang, including the creation of an expanding, hot, dense, almost-perfectly uniform Universe.
- Provide a mechanism for explaining those three puzzles — the temperature uniformity, the lack of high-energy relics, and the flatness problem — that the Big Bang has no solution for.
- Finally, and perhaps most importantly it must make new, testable predictions that are different from the standard Big Bang that it’s attempting to supersede.
The idea of inflation, and the hope that it could do so, began in late 1979, when Alan Guth wrote the idea down in his notebook.

What inflation specifically hypothesized is that the Big Bang wasn’t the beginning, but rather was set up by a prior stage of the Universe. In this early state — dubbed an inflationary state by Guth — the dominant form of energy wasn’t in matter or radiation, but was inherent to the fabric of space itself, and possessed a very large energy density.
This would cause the Universe to expand both rapidly and relentlessly, driving any pre-existing matter apart. The Universe would be stretched so large it would be indistinguishable from flat. All the parts that an observer (like us) would be able to access would now have the same uniform properties everywhere, since they originated from a previously-connected state in the past. And since there would be a maximum temperature the Universe achieved when inflation ended, and the energy inherent to space transitioned into matter, antimatter, and radiation, we could avoid the production of leftover, high-energy relics.

All at once, all three of those puzzles that the Big Bang couldn’t explain were solved. This was truly a watershed moment for cosmology, and immediately led to a deluge of scientists working to correct Guth’s original model in order to reproduce all of the Big Bang’s successes. Guth’s idea was published in 1981, and by 1982, two independent teams — Andrei Linde and the duo of Paul Steinhardt and Andy Albrecht — had done it.
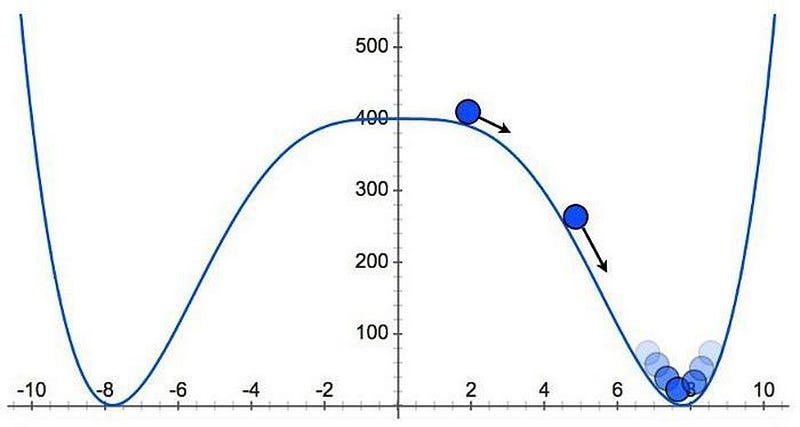
The key was to picture inflation as a slowly-rolling ball atop a hill. As long as the ball remained atop the plateau, inflation would continue to stretch the fabric of space. But when the ball rolls down the hill, inflation comes to an end. As the ball rolls into the valley below, energy inherent to space gets transferred into matter, antimatter and radiation, leading to a hot Big Bang, but with a finite temperature and energy.
At last, not only did we have a solution to all of the problems that the Big Bang couldn’t resolve, but we could reproduce all of its successes. The key, then, would be to make new predictions that could then be tested.
The 1980s were full of such predictions. Most of them were very general, occurring in practically all viable models of inflation that one could construct. In particular, we realized that inflation had to be a quantum field, and that when you have this rapid, exponential expansion occurring with an extremely high energy inherent to space itself, these quantum effects can have impacts that translate onto cosmological scales.
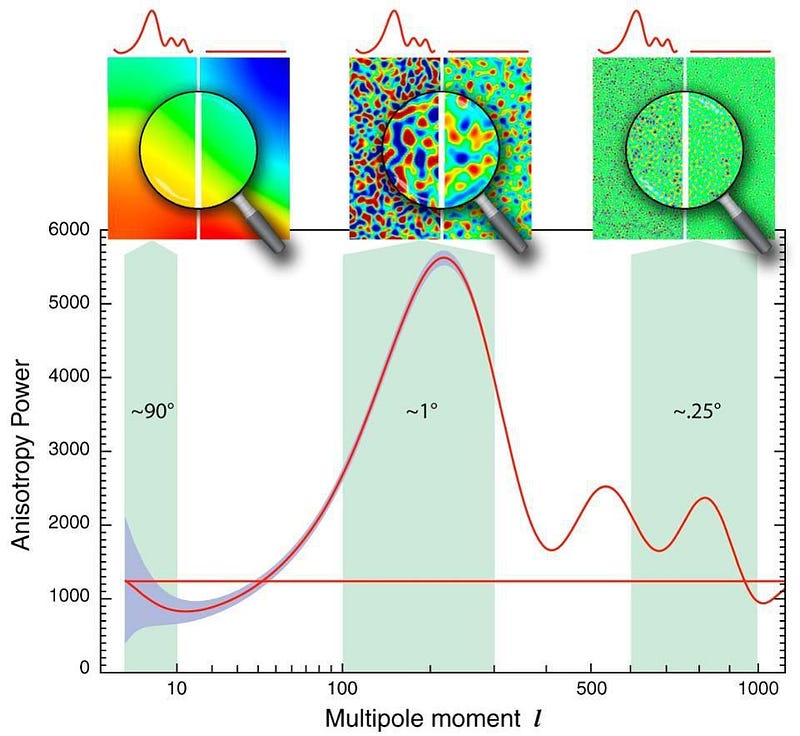
In brief, the six most generic predictions were:
- There should be an upper-limit to the maximum temperature the Universe achieves post-inflation; it cannot approach the Planck scale of ~10¹⁹ GeV.
- Super-horizon fluctuations, or fluctuations on scales larger than light could have traversed since the Big Bang, should exist.
- The quantum fluctuations during inflation should produce the seeds of density fluctuations, and they should be 100% adiabatic and 0% isocurvature. (Where adiabatic and isocurvature are the two allowed classes.)
- These fluctuations should be almost perfectly scale-invariant, but should have slightly greater magnitudes on larger scales than smaller ones.
- The Universe should be nearly, but not quite, perfectly flat, with quantum effects producing curvature only at the 0.01% level or below.
- And the Universe should be filled with primordial gravitational waves, which should imprint on the cosmic microwave background as B-modes.
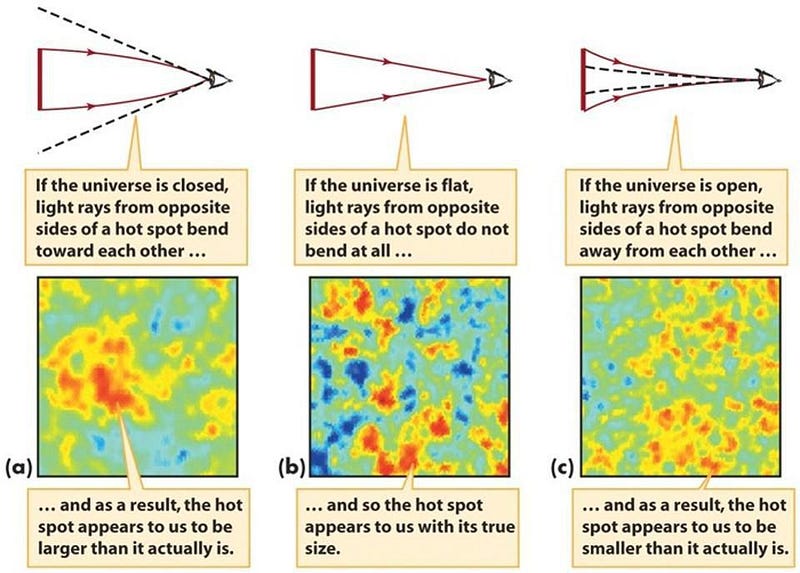
It’s now 2019, and the first four predictions have been observationally confirmed. The fifth has been tested down to the ~0.4% level and is consistent with inflation, but we haven’t reached the critical level. Only the sixth point has not been tested at all, with a famous false-positive detection appearing earlier this decade owing to the BICEP2 collaboration.
The maximum temperature has been verified, by looking at the cosmic microwave background, to be no greater than about 10¹⁶ GeV.
Super-horizon fluctuations have been seen from the polarization data provided by both WMAP and Planck, and are in perfect agreement with what inflation predicts.
The latest data from structure formation indicates that these early, seed fluctuations are at least 98.7% adiabatic and no more than 1.3% isocurvature, consistent with inflation’s predictions.
But the best test — and what I’d call the most significant confirmation of inflation — has come from measuring the spectrum of the initial fluctuations.
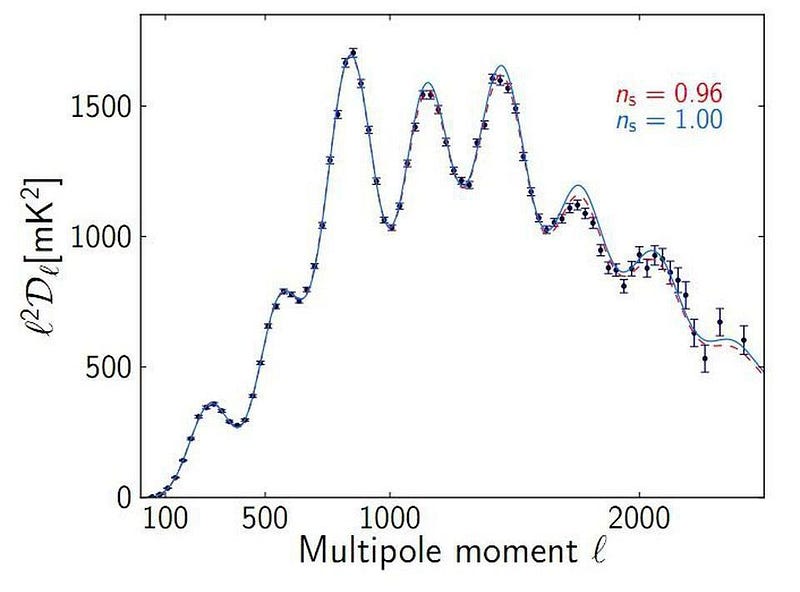
Inflation is very particular when it comes to what sorts of structure should form on different scales. We have a quantity that we use to describe how much structure forms on large cosmic scales versus smaller ones: n_s. If you formed the same amount of structure on all scales, n_s would equal 1 exactly, with no variations.
What inflation generically predicts, however, is that we will have an nsthat’s almost, but slightly less than, 1. The amount we depart from 1 by is determined by the specific inflationary model. When inflation was first proposed, the standard assumption was that n_s would be exactly equal to 1. It wouldn’t be until the 2000s that we became capable of testing this, through both the fluctuations in the cosmic microwave background and the signature of baryon acoustic oscillations.
As of today, n_s is approximately 0.965 or so, with an uncertainty of around 0.008. This means there’s about a 4-to-5 sigma certainty that n_s is truly less than 1, a remarkable confirmation of inflation.
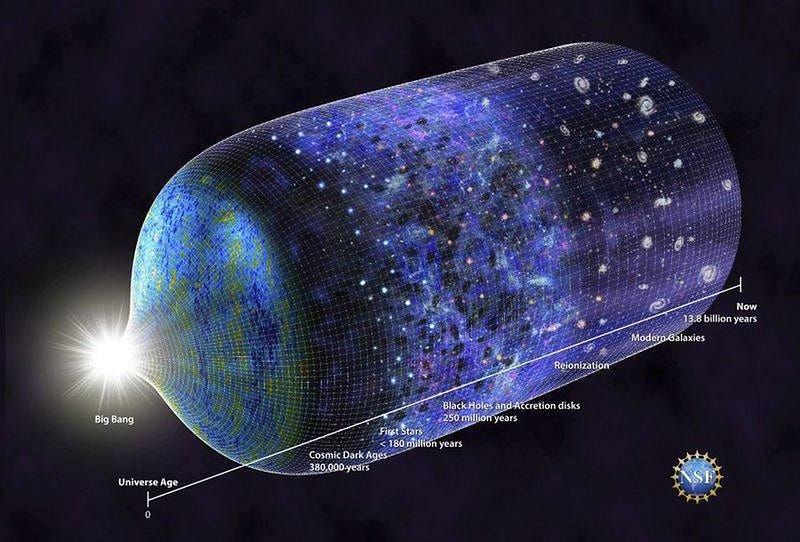
The Big Bang became our theory of the Universe when the leftover glow was discovered in the form of the cosmic microwave background. As early as 1965, the critical evidence had come in, enabling the Big Bang to succeed where its competitors failed. Over the subsequent years and decades, measurements of the cosmic microwave background’s spectrum, the abundance of the light elements, and the formation of structure only strengthened the Big Bang. Although alternatives persist, they cannot stand up to the scientific scrutiny that the Big Bang does.

Inflation has literally met every threshold that science demands, with clever new tests becoming possible with improved observations and instrumentation. Whenever the data has been capable of being collected, inflation’s predictions have been verified. Although it’s perhaps more palatable and fashionable to be a contrarian, inflation is the leading theory for the best reason of all: it works. If we ever make a critical observation that disagrees with inflation, perhaps that will be the harbinger of an even more revolutionary theory of how it all began.
Send in your Ask Ethan questions to startswithabang at gmail dot com!
Ethan Siegel is the author of Beyond the Galaxy and Treknology. You can pre-order his third book, currently in development: the Encyclopaedia Cosmologica.